The first non-opoid painkiller

In the nineteenth century, the invention of anesthesia was considered a gift from God. But post-operative pain relief has continued to rely on opioids, derivatives of opium, the addictive substance employed since ancient times. Although no other drug has managed to match the rapid, potent, and broadly effective relief delivered by opioids, their side effects have led to decades of addiction and overdose, leaving researchers keen to find a better solution.
This all changed in January 2025, when the FDA approved Vertex Pharmaceuticals’s Journavx(suzetrigine): thefirst non-opioid pain reliever suitable for treating post-surgery pain. Clinical trials found no signs of the problematic side effects associated with opioids: no drug abuse, tolerance, or withdrawal. But this was not an easy win: Vertex and other pharma companies spent decades searching for drugs like this to no avail.
Opioids are used primarily to treat nociceptive pain, pain caused by tissue damage from injury or disease. This damage activates nearby nociceptors: sensory neurons that signal physical or chemical harm. These nociceptors send signals up to the central nervous system – the brain and spinal cord – and the brain then creates a localized sensation of pain, drawing your attention to the threat.
Traditional opioids mimic opium, a compound found in the poppy plant that contains morphine. Opioids alleviate pain by acting on one of the three main opioid receptors, mu (μ) opioid receptors, which are distributed throughout the central nervous system, particularly in the brain. When opioids bind to the brain’s mu receptors, this suppresses incoming pain signals from the damaged site’s nociceptors, preventing the brain from creating the sensation of pain even when tissue damage is present.
Our bodies naturally produce their own opioids – such as endorphins, endogenous morphine – to briefly blunt pain during moments of stress or injury. However, these are far weaker and shorter-acting than prescription opioids since they degrade quickly, remain localized, and are released in short, controlled bursts. Prescription opioids, on the other hand, flood the brain with higher doses that linger for hours.
Crucially, opioids don’t just kill pain: they also incite pleasure. When the mu opioid receptors present in the reward center of the brain are activated, this reduces the secretion of a neurotransmitter called GABA, which works to inhibit dopamine-producing neurons. As GABA release declines, dopamine spikes, lighting up the reward center and inducing pleasure.
With the body’s natural opioids, this is fleeting and unproblematic. When properly prescribed, even synthetic opioids are no issue for most patients: under severe post-surgical pain, opioids mostly function to normalize disrupted brain function, dampening any pleasurable effect. But for some, whether due to genetics or inappropriate administration (e.g. a prescription that goes on after the pain’s source has been relieved), the intensity of prescription opioids produces a prolonged dopamine spike, along with a marked sensation of euphoria: a recipe for addiction.
With chronic use, the body’s natural opioid system becomes dysfunctional. Fewer natural opioids are produced and opioid receptors become desensitized. As a result, the patient develops a tolerance, requiring higher and higher doses to even feel normal.
The nineteenth century witnessed the creation of morphine, codeine, and heroin (which was sold over-the-counter), as well as the invention of the hypodermic syringe. By the turn of the century, 15 percent of all prescriptions dispensed in Boston were for opioids, which were used for everything from menstrual cramps to children’s coughs, and as many as 300,000 Americans, or 0.5 percent of the population, were opiate addicts. Anti-narcotics laws proliferated throughout the states, and the medical community expressed concerns about the liberal provision of addictive drugs. These mounting pressures led to the passage of the Harrison Narcotic Act in 1914, which made opium and opiates the first regulated substances in the United States.
Peripheral solutions
Unlike opioids, which act within the central nervous system, Journavx does not meaningfully interact with the brain. Instead, it targets a specific sodium ion channel found almost exclusively on peripheral nociceptors, the pain-sensing neurons throughout your body. Ion channels, whether sodium, potassium, or calcium, are like tiny doors embedded on the neuron’s membrane: when a door opens, ions rush in or out and the neuron fires, sending an electrical signal to the next cell.
Three sodium channels are found primarily on nociceptors: NaV1.7, NaV1.8, and NaV1.9. Suzetrigine selectively blocks NaV1.8, which stops nociceptors from sending pain signals tothe brain. Rather than preventing your brain from receiving pain signals, as opioids do, it prevents your neurons from transmitting them. In essence, Journavx works from the bottom up to alleviate pain, rather than the top down.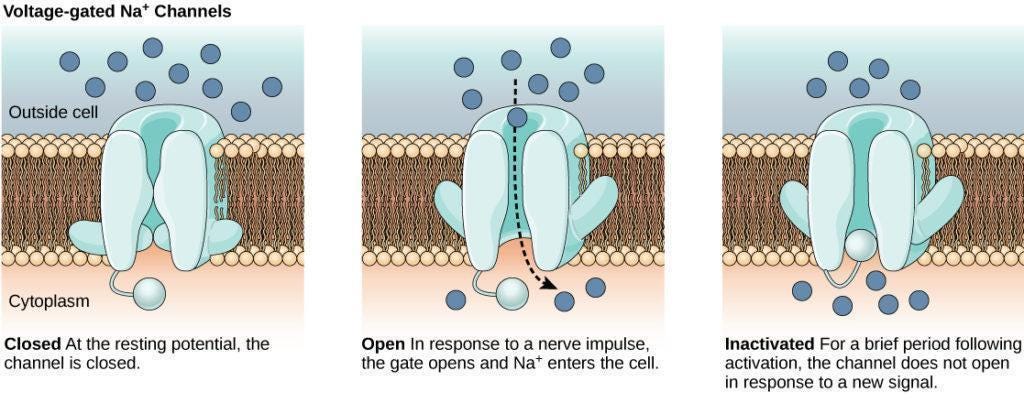
Critically, the NaV1.8 channel is largely absent from the central nervous system. This means that suzetrigine does not affect the brain, which means users do not experience the same euphoria that is triggered by opioids. This prevents addiction and abuse, as well as the depressive effects on breathing or heart rate typical with opioids.
At first glance, this may seem like a straightforward solution, especially given the urgent demand for non-opioid alternatives. So why did it take so long?
Unlike diseases with well-defined biological causes, pain is a broad symptom rooted in complex and overlapping pathways. Many of these are deeply intertwined with vital bodily functions like blood pressure, immune response, and respiration. Together, this makes it difficult to isolate a target that can be blocked without collateral damage.
A particularly good example of this predicament involves TRPV1, also known as the capsaicin receptor. It is an ion channel mainly found in nociceptors, and is responsible for the pain you feel when eating spicy foods. In clinical trials, TRPV1 inhibitors effectively alleviated pain, but researchers found that they unexpectedly disrupted thermoregulation, causing patients to experience hyperthermia, or overheating, with one trial participant sustaining a 104 degrees fahrenheit fever for hours.
Another example involves nerve growth factor inhibitors like tanezumab. Although tanezumab alleviated inflammatory pain from conditions like osteoarthritis, Phase III trials revealed an unfortunate side effect: rapidly progressive osteoarthritis. Researchers hypothesized that because patients felt so much better, they overused their arthritic joints, accelerating damage. Although further trials were conducted at lower doses and with restrictions, the FDA ultimately voted against its approval. Tanezumab’s story reflects a difficulty in developing painkillers: while pain can cause excessive suffering, it also serves as a vital warning sign that must be selectively maintained.
Men on fire
Vertex has historically focused on developing drugs targeting ion channels. These channels play a major role in cellular signaling, meaning that compounds that act upon them can produce large, rapid physiological effects. Ion channels are ‘really good drug targets’, Paul Negulescu, head of Vertex's pain program, says, ‘They just require a lot of care and attention to how you measure them’.
The discovery of the NaV sodium channels, made independently in the early 2000s by two different researchers, opened a new frontier in pain research. Both observed that mutations affecting NaV1.7 caused abnormalities in the experience of pain, a major clue that pain might be mediated through that specific sodium channel.
Stephen Waxman, a professor of neurology, neuroscience, and pharmacology at Yale’s medical school, discovered that a community in Alabama had numerous individuals suffering from erythromelalgia or ‘Man on Fire’ syndrome. These individuals experienced mild warmth – such as from wearing a sweater or shoes – as intense burning pain. Waxman’s research tied this phenomenon to mutations in the SCN9A gene, which is involved in the production of NaV1.7 channels. Meanwhile, Geoff Woods, a clinical geneticist at St. James’s University Hospital in Leeds, uncovered a complementary discovery. He observed congenital insensitivity to pain within specific Pakistani communities, also tracing it back to mutations in the SCN9A gene.
This congenital insensitivity provided a particularly compelling genetic validation for a drug target, as the affected individuals were entirely normal except for their inability to feel pain, unlike prior similar cases. Related channels like NaV1.8 and NaV1.9 were also investigated by Woods’s team and found relevant for pain signaling.
But despite the initial enthusiasm surrounding these discoveries, researchers soon encountered significant obstacles: NaV1.7 inhibitors failed to relieve pain during clinical trials. Researchers eventually uncovered that the congenital absence of NaV1.7 did not eliminate pain signals but instead amplified the production of natural painkillers called enkephalins. They concluded that completely blocking the channel, which would be required to replicate this effect pharmaceutically, was impractical.
So researchers turned their attention to the other promising sodium channel: NaV1.8. Again, research began with setbacks: in 2015, it was discovered that individuals with Brugada syndrome, a disorder characterized by abnormal heart rhythms and sudden cardiac death, also had mutations in the gene encoding NaV1.8.
Despite this challenge, researchers still thought NaV1.8 had potential. Woods’ research genetically validated it, showing that mutations in NaV1.8 affect pain signalling. Researchers at the University of Alcalá confirmed that mice genetically engineered to lack Nav1.8 channels showed virtually no spontaneous nerve activity after injury – activity thought to underlie certain chronic pains. Additionally, NaV1.8's almost exclusive presence in the peripheral nervous system (rather than in the brain) suggested that it might uniquely limit undesirable central side effects.
Ion channels
As Vertex researchers searched for NaV1.8 inhibitors, they made use of Negulescu’s E-VIPR technology, which enabled them to conduct more than 50,000 tests per day to identify compounds that blocked NaV1.8 without affecting other ion channels. This was essential because the human body contains nine known voltage-gated sodium channel types, each with a distinctive ‘personality’ – a unique pattern of rapid opening, closing, and voltage sensitivity – making high throughput key to pinpointing an appropriately selective drug.
But even with this tool, Negulescu described the iterative learning process as ‘painful’. Vertex spent a decade screening millions of compounds before finding a promising class of molecules. Another decade was spent on optimization, conducting tens of thousands of screenings to maximize potency and selectivity (a drug is selective if it binds only to the target proteins and nowhere else).
Vertex faced several failures in preclinical and clinical testing. Between 2018 and 2022, they terminated development for three generations of NaV1.8 inhibitors, VX-150, VX-128, and VX-961, due to dosing and tolerability issues. However, unlike previous attempts with NaV1.7, TRPV1, and nerve growth factor inhibitors, the pathway overall did not exhibit fatal flaws, and so research continued.
Eventually, this iterative process produced VX-548, which was discovered to be many times more selective and potent than earlier candidates. In 2022, two Phase II proof-of-concept studies yielded positive results. In 2024, Phase III trials validated VX-548’s efficacy in treating acute pain with minimal adverse effects. During this process, the FDA granted VX-548, now suzetrigine, Fast Track and Breakthrough Therapy designations, processes designed to accelerate the development and review of crucial pharmaceutical innovations.
On July 30, 2024, the FDA accepted Vertex’s New Drug Application, filing it under priority review. Exactly six months later, on January 30, 2025, it was approved, marking suzetrigine – sold under the brand name Journavx – as the first non-opioid analgesic for treating acute pain.
Pain points
Journavx isn’t a silver bullet. It has not yet been tested or approved for treating chronic pain, from which over 20 percent of Americans suffer. Across its clinical trials, 85 and 98 percent of participants were female. This reflects a broader pattern in painkiller trials, which often rely on surgical models like bunionectomy and abdominoplasty (‘tummy tuck’) – procedures overwhelmingly performed on women. Since 2022, the bipartisan ‘No Pain Act’ has mandated Medicare and other government health plans to cover this class of medication in outpatient surgical settings, private insurance coverage is still in flux: without insurance, a week’s worth of Journavx costs around $230, compared to $10–20 for a low-dose opioid-acetaminophen combination medication.
Journavx failed to outperform that opioid-acetaminophen combination in clinical trials. Todd Bertoch, an anesthesiologist involved in suzetrigine’s Phase III trials, explains that the drug likely won’t serve as an outright opioid replacement, but the first step on the journey to minimizing opioid usage. If paracetamol and ibuprofen are inadequate for pain relief, Journavx can now be prescribed as the next alternative treatment, instead of mild- to moderate-strength opioids.
It will almost certainly improve: Vertex’s scientists are continuing their decades-long project to iterate and screen for even more potent and selective NaV1.8 blockers. They are also investigating complementarities with NaV1.7 inhibitors. A Phase III clinical trial of suzetrigine for diabetic peripheral neuropathy, which involves chronic pain, is currently underway.
Journavx is the product of 27 years, billions of dollars, millions of molecules screened, dozens of monkeys and rats and data from over 2,400 surgical patients, all distilled into a single 50-mg blue tablet.
Vertex chose to keep funding and pushing forward through decades of work that industry professionals describe as ‘tedious’, ‘mind-numbing’, and ‘painstaking’, a slog driven by slow, incremental progress and frequent setbacks. In exchange, humanity now has its first non-opioid painkiller.
Michelle Ma studies economics at the University of Chicago.
Thanks for reading The Works in Progress Newsletter! Subscribe for free to receive new posts.
What's Your Reaction?
 Like
0
Like
0
 Dislike
0
Dislike
0
 Love
0
Love
0
 Funny
0
Funny
0
 Angry
0
Angry
0
 Sad
0
Sad
0
 Wow
0
Wow
0